All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the International Myeloma Foundation or HealthTree for Multiple Myeloma.
The Multiple Myeloma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Multiple Myeloma Hub cannot guarantee the accuracy of translated content. The Multiple Myeloma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Multiple Myeloma Hub is an independent medical education platform, sponsored by Bristol Myers Squibb, GSK, Johnson & Johnson Innovative Medicine, Legend Biotech, Pfizer, Roche, and Caribou Biosciences. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View multiple myeloma content recommended for you
 Rafael Fonseca
Rafael FonsecaMultiple myeloma (MM) is characterized by considerable clonal genetic heterogeneity. Precursor stages, such as monoclonal gammopathy of undetermined significance and smoldering MM, have also demonstrated considerable heterogeneity (Figure 1).
Figure 1. Clonal heterogeneity in MM and precursor stages*
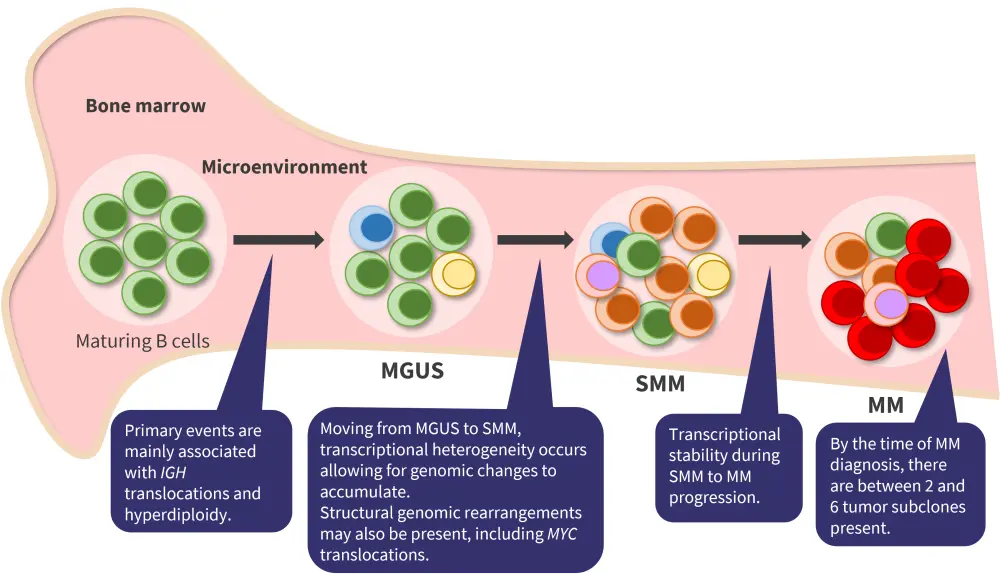
MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; SMM, smoldering multiple myeloma.
*Adapted from Dutta et al.1 and Aksenova et al.2
This clonal heterogeneity presents a great challenge for treatment, as a malignancy in no two patients may have the exact same genetic makeup. However, this also presents an opportunity to assess treatments targeted to particular genetic subgroups. Precision medicine has succeeded in solid tumors, e.g., with the use of HER2-targeted therapy in breast cancer, but will it be possible in MM treatment?1 In a recent article published in Frontiers in Oncology, Darren Pan and Joshua Ricter3 discussed the current landscape for precision medicine in MM.
Current targeted treatments
t(11;14)
Currently, there are no therapies approved by the U.S. Food and Drug Administration (FDA) for a particular mutational subset in MM. However, interesting data has been seen for the use of venetoclax in clinical trials and real-world settings. Patients with MM and the presence of t(11;14) appear to respond to a greater extent to treatment with venetoclax compared with patients without this translocation. t(11;14) is detected in 15–20% of patients with MM, and it is associated with a higher expression of and dependency on BCL-2.4
In the phase III Bellini trial (NCT02755597), patients with relapsed/refractory (RR)MM (N = 291) were treated with bortezomib and dexamethasone with either venetoclax or a placebo.3 While a median progression-free survival of 22.4 months was achieved in the venetoclax arm compared with 11.5 months in the placebo arm, there was an increase in adverse events. Grade ≥3 neutropenia (18% vs 7%) and pneumonia (16% vs 9%) were elevated in the venetoclax arm compared with the placebo arm. This led to a decreased overall survival due to infectious complications and study discontinuation. In spite of this, it was noted that patients harboring t(11;14) attained superior responses with a median progression-free survival not yet reached vs 9.9 months in the placebo arm, but without the increased treatment-related deaths.3
While other studies have shown similar results demonstrating patients with t(11;14) doing better than patients without the translocation, no standard dose has been used, with some reports noting up to 800 mg and others 400 mg or lower. As a result, current studies examining venetoclax in combination with backbone therapies are now enrolling only patients that possess the t(11;14) translocation to investigate its efficacy and safety further in this subgroup.
Why assessing t(11;14) can be the first step towards personalized therapy in MM?
Future possible targets3
New precision medicine approaches are being investigated beyond the presence of t(11;14). Genome sequencing and new -omics are accelerating the understanding of the particularities of MM biology and helping to discover new biomarkers and target candidates.3 Below, we have listed some of the latest identified targetable mutations that could drive the future of personalized medicine in MM.
FGFR3 inhibition
FGFR3 can become dysregulated along with WHSC1/MMSET in patients with MM who harbor the t(4;14)(p16.3;q32) translocation, which is found in ≤20% of newly diagnosed patients. This translocation is a poor prognostic marker in these patients.
Inhibiting FGFR3 has been investigated clinically with non-selective multikinase inhibitors, with little success. Specific FGFR3 inhibitors, such as erdafitinib, which target the pathway specifically, may be effective. The Myeloma-Developing Regimens Using Genomics (MyDRUG) trial (NCT03732703) will test erdafitinib with ixazomib and pomalidomide in patients with FGFR3-activating mutations.
CDK4/6 inhibition
Cyclin-dependent kinase (CDK)4/6 controls transition from the G1 to the S phase of the cell cycle. CDKN2C, an endogenous inhibitor of CDK4/6, may be decreased in MM and lead to increased proliferation. This deletion is detected in 15% of patients with MM and is associated with decreased overall survival.
In vitro studies with abemaciclib have shown inhibition of MM cell growth and cytotoxicity dependent on the dose. Abemaciclib will also be used in the MyDRUG trial for patients with CDKN2C loss along with ixazomib and pomalidomide.
Another CDK4/6 inhibitor that has been assessed in vitro is palbociclib. By itself, palbociclib produces cytostatic effects on MM cell lines, but in combination with dexamethasone and bortezomib it can enhance cytotoxicity. In a phase I/II trial that did not identify biomarkers, five out of 25 patients achieved an overall response rate, and the study failed to proceed to the next stage.
Mutant IDH inhibition
Isocitrate dehydrogenase (IDH) mutations are uncommon in MM at only 0.5%, compared with ~16% in acute myeloid leukemia. Nonetheless, patients with mutant IDH2 will be treated with enasidenib-based combinations in the MyDRUG trial.
SETD2 methyltransferase inhibition
As a histone methyltransferase, SETD2 is involved in DNA repair, alternative splicing, and transcriptional silencing. SETD2 mutations are often found in MM, especially in RR disease. In vivo studies have shown that SETD2 inhibitors suppress proliferation in MM cell lines and xenograft mouse models. There is currently one ongoing phase I trial for a SEDT2 inhibitor in patients with MM.5
NTRK inhibition
Neurotrophin receptor tyrosine kinase (NTRK) fusions are commonly found in certain solid tumors but are only present in <1% of MM cases. However, larotrectinib, a NTRK inhibitor, is being investigated in the MATCH trial (NCT02465060) for both solid and hematologic malignancies, including MM.
Mcl-1 inhibition
The anti-apoptotic protein Mcl-1 is involved in myeloma cell survival. Approximately 40% of patients with MM have changes on chromosome 1q21, where the MCL-1 gene resides. These amplifications or gains at 1q21 are associated with significantly reduced survival times.
Venetoclax resistance frequently involves MCL-1 upregulation, and therefore, there is speculation over whether Mcl-1 and Bcl-2 inhibition would produce effective clinical synergy. AMG 176, an Mcl-1 inhibitor, has shown the ability to cause apoptosis in blood cancer cell lines when given with carfilzomib and dexamethasone. This combination is being evaluated in an ongoing phase I trial in patients with RRMM. Another Mcl-1 inhibitor, AZD5991, is also being evaluated in a phase I study in MM and acute myeloid leukemia with or without venetoclax.
MDM2 inhibition
Ubiquitination of the tumor suppressor p53 is controlled by mouse double minute 2 (MDM2), resulting in its degradation. MM cells can reduce the expression of p53 in a number of different ways, including MDM2 overexpression, which then leads to increased cell division. Nutlin is an MDM2 inhibitor that has promoted apoptosis in MM cell lines and shown synergy in a preclinical setting with bortezomib. However, synergy was only seen in MM cells with wild type p53; therefore, treatment with MDM2 inhibitors has the potential to select for p53-mutated cells.
Two MDM2 inhibitors are currently being evaluated. KRT-232 in combination with carfilzomib, lenalidomide, and dexamethasone is being assessed in a phase I trial in patients with RRMM (NCT03031730). Idasanutlin is also being assessed in a phase I/II trial (NCT02633059) with ixazomib and dexamethasone in patients with RRMM who have the del(17p) or monosomy 17.
Conclusion
The field of precision medicine is still very young in MM, but there are a number of promising agents moving from preclinical studies into clinical trials currently. In addition, the positive real-world results from venetoclax in patients with the t(11;14) translocation may indicate this could be the first precision medicine used successfully in MM, although further testing is ongoing. The MyDRUG trial should provide a large amount of data on personalized therapy and is greatly anticipated. In any case, the significant degree of clonal heterogeneity in MM remains a challenge and will probably require targeted and non-targeted agents to be used together in appropriate combinations.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content